

Loss of ALS-associated TDP-43 in zebrafish causes muscle degeneration, vascular dysfunction, and reduced motor neuron axon outgrowth
Proc Natl Acad Sci U S A. 2013 Mar 1. [Epub ahead of print]
| Authors/Editors: |
Schmid B Hruscha A Hogl S Banzhaf-Strathmann J van der Zee J Teucke M Eimer S Hegermann J Kittelmann M Kremmer E Cruts M Solchenberger B van Bebber F Van Broeckhoven C Edbauer D Lichtenthaler SF Haass C |
|---|---|
| Publication Date: | 2013 |
| Type of Publication: | Journal Article |
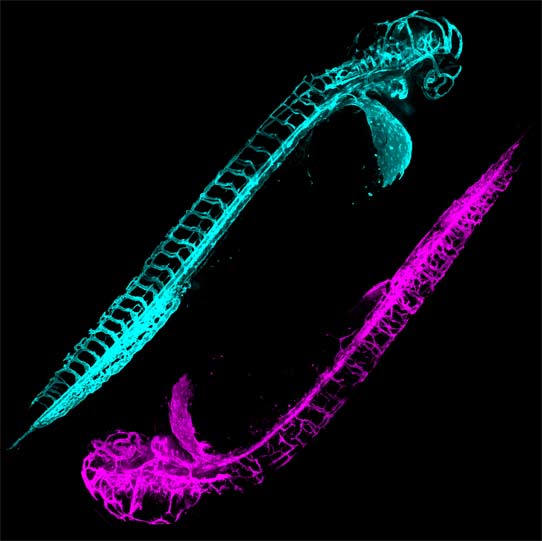
ABSTRACT:
Mutations in the Tar DNA binding protein of 43 kDa (TDP-43; TARDBP) are associated with amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration with TDP-43+ inclusions (FTLD-TDP). To determine the physiological function of TDP-43, we knocked out zebrafish Tardbp and its paralogue Tardbp (TAR DNA binding protein-like), which lacks the glycine-rich domain where ALS- and FTLD-TDP-associated mutations cluster. tardbp mutants show no phenotype, a result of compensation by a unique splice variant of tardbpl that additionally contains a C-terminal elongation highly homologous to the glycine-rich domain of tardbp. Double-homozygous mutants of tardbp and tardbpl show muscle degeneration, strongly reduced blood circulation, mispatterning of vessels, impaired spinal motor neuron axon outgrowth, and early death. In double mutants the muscle-specific actin binding protein Filamin Ca is up-regulated. Strikingly, Filamin C is similarly increased in the frontal cortex of FTLD-TDP patients, suggesting aberrant expression in smooth muscle cells and TDP-43 loss-of-function as one underlying disease mechanism.

