Generation and deposition of Aβ43 by the virtually inactive presenilin-1 L435F mutant contradicts the presenilin loss-of-function hypothesis of Alzheimer’s disease
EMBO Mol Med. 2016 May 2;8(5):458-65
| Authors/Editors: |
Benedikt Kretner Johannes Trambauer Janina Mielke Peer-Hendrik Kuhn Elisabeth Kremmer Armin Giese Stefan Lichtenthaler Christian Haass Thomas Arzberger Harald Steiner |
|---|---|
| Publication Date: | 2016 |
| Type of Publication: | Journal Article |
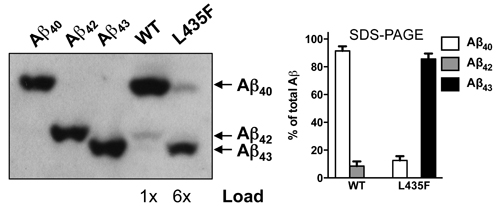
As stated by the prevailing amyloid cascade hypothesis, Alzheimer’s disease (AD) is caused by the aggregation and cerebral deposition of long amyloid-β peptide (Aβ) species, which are released from a C-terminal amyloid precursor protein fragment by c-secretase. Mutations in its catalytic subunit presenilin-1 (PS1) increase the Aβ42 to Aβ40 ratio and are the major cause of familial AD (FAD). An opposing hypothesis states that loss of essential presenilin functions underlies the disease. A major argument for this hypothesis is the observation that the nearly inactive PS1 L435F mutant, paradoxically, causes FAD. We now show that the very little Aβ generated by PS1 L435F consists primarily of Aβ43, a highly amyloidogenic species which was overlooked in previous studies of this mutant. We further demonstrate that the generation of Aβ43 is not due to a trans-dominant effect of this mutant on WT presenilin. Furthermore, we found Aβ43-containing plaques in brains of patients with this mutation. The aberrant generation of Aβ43 by this particular mutant provides a direct objection against the presenilin hypothesis.