

Content
Frontotemporal lobar degeneration (FTLD)
Frontotemporal lobar degeneration (FTLD) is the second most common form of dementia under the age of 65. FTLD is characterized by progressive changes in social behavior, personality and/or language dysfunction and a selective loss of neurons in the temporal and frontal cortex. There are two major pathological subtypes in FTLD. About 40% of FTLD patients are pathologically characterized by inclusions of hyperphosphorylated tau (FTLD-tau). The most common FTLD variant presents with TAR DNA-binding protein 43 (TDP-43) inclusions (FTLD-TDP), and about 10% of FTLD cases contain deposits of the Fused in sarcoma (FUS) protein.
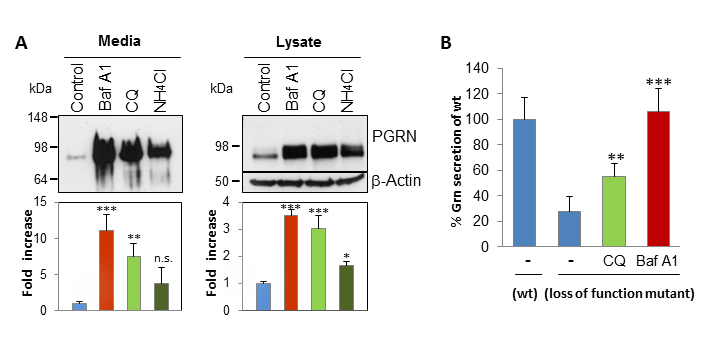
Figure 1: Alkalizing reagents increase intracellular and secreted GRN levels and might be promising candidates for a therapy approach.
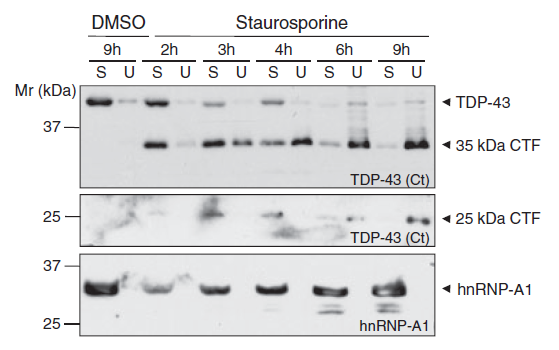
Figure 2: Caspase-dependent cleavage of TDP-43 leads to the formation of insoluble TDP-43 fragments.
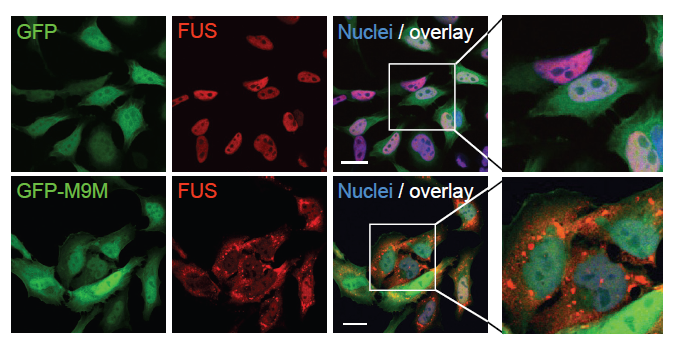
Figure 3: FUS localizes to cytoplasmic stress granules after inhibition of the Transportin pathway.
Clinically, FTLD is related with amyotrophic lateral sclerosis (ALS), the most frequent motor neuron disease, in which degeneration of motor neurons leads to progressive muscle weakening and eventually fatal paralysis. Since up to 50% of ALS patients develop cognitive impairment and FTLD patients also can present with motor neuron disease, it has long been recognized that the two diseases are related. This idea has been strengthened by the recent discovery that the two diseases share as a key characteristic the accumulation of the TDP-43 or the FUS protein within abnormal protein aggregates.
TDP-43 and FUS are normally localized in the nucleus, where they bind DNA/RNA and regulate transcription and RNA splicing. Since pathogenic TDP-43 and FUS inclusions are mostly observed in the cytosol and and inclusion-bearing cells often show a loss or reduction of nuclear staining, it is thought that the loss of nuclear TDP-43 and FUS and/or an excessive accumulation in the cytosol are involved in neuronal toxicity. It is still unclear how cytoplasmic TDP-43 and FUS positive inclusions arise and how they are linked to neuronal cell death and pathogenesis.
Genetic linkage studies and/or mutation screenings identified autosomal dominant mutations in the progranulin gene (GRN) accounting for about 20% of familiar FTLD-TDP cases. Of the approximately 70 mutations reported to date, most are loss-of-function mutations leading to GRN haploinsufficiency, which results in a severe reduction of GRN levels in tissues and biological fluids of patients. Additionally, missense mutations might lead to folding defects aberrant processing and consequently result in reduced secretion.
Since GRN mutations account only partially for familial FTLD-TDP cases and carriers of identical mutations show a high variability in age of onset and in pathological presentation additional genetic factors or environmental influences were postulated to play a role for manifestation of the disease. Consistent with that hypothesis, the first genome-wide association study (GWAS) in FTLD with TDP-43 inclusions identified three single nucleotide polymorphisms (SNPs) at the TMEM106B gene locus on chromosome 7p21.3 as a risk factor. TMEM106B variants specifically increase the risk for FTLD-TDP in patients with mutations in GRN.
GRN is a secreted protein, which serves as a wound healing and growth factor and has neurotrophic properties. Since all mutations identified so far lead to a severe reduction in GRN, it is thought that reduced GRN levels are causally linked to neurodegeneration. It is still unclear how loss of GRN leads to the formation of TDP-43 positive inclusions and a selective loss of neurons in the frontal and temporal cortex.
To gain insights into the molecular mechanisms leading to TDP-43 and FUS inclusion formation and neurodegeneration, we study the cell biology of TDP-43, FUS and GRN. Since cytosolic mislocalization of the nuclear proteins TDP-43 and FUS is a key feature in FTLD/ALS, we aim to understand nucleocytoplasmic transport of these RNA/DNA binding proteins. Furthermore, we study the association of FUS and TDP-43 with stress granules, since our previous work has implicated stress granules in FUS/TDP-43 pathology.
In order to understand the link between GRN and TDP-43 pathology, we study the cell biology of GRN with the main focus on regulation of GRN expression levels. We furthermore elucidate the potential of stimulation of GRN expression for therapy of FTLD-TDP. Since TMEM106B is a largely uncharacterized protein, we study the basic cell biology and biochemistry of TMEM106B to shed light on its biological function, with the aim of understanding how TMEM106B affects TDP-43 pathology in a GRN dependent manner.
Responsible for content: Prof. Dr. Dr. h.c. Christian Haass; Dr. Dorothee Dormann; Dr. Anja Capell